This functions takes an extracted MS/MS spectra and writes it to a .msp
file format. This function incorporates the extracted MS/MS spectra along
with metadata for the compound.
Arguments
- spec
a data frame containing the extracted MS/MS spectra, the following columnS are required:
- mz_precursor
- rt
- mz
- intensity
- spec_metadata
a data frame containing the values to be including in the resulting
.mspfile. The following columns are required as vital information for the.mspoutput.- NAME
- PRECURSORTYPE
- FORMULA
- RETENTIONTIME
- IONMODE
The following fields can be included in the resulting
.mspfile, but are not required to be present in the metadata table. If the column does not exist in the column or the value is missing, a blank field will be exported- INCHIKEY
- SMILES
- CCS
- COLLISIONENERGY
- INSTRUMENTTYPE
- msp_name
a string with the name of the
.mspfile excluding the file extension
Examples
# Example with single MS/MS spectra -----
# Importing the Spectrum of Procyanidin A2 in negative ionization mode
# and 20 eV as the collision energy
ProcA2_file <- system.file("extdata",
"ProcyanidinA2_neg_20eV.mzXML",
package = "MS2extract"
)
# Region of interest table (rt in seconds)
ProcA2_data <- data.frame(
Formula = "C30H24O12", Ionization_mode = "Negative",
min_rt = 163, max_rt = 180
)
ProcA2_raw <- import_mzxml(ProcA2_file, ProcA2_data)
#> • Processing: ProcyanidinA2_neg_20eV.mzXML
#> • Found 1 CE value: 20
#> • Remember to match CE velues in spec_metadata when exporting your library
#> • m/z range given 10 ppm: 575.11376 and 575.12526
# Extracting the most intense MS2 scan
ProcA2_ext <- extract_MS2(ProcA2_raw)
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Warning: `position_stack()` requires non-overlapping x intervals.
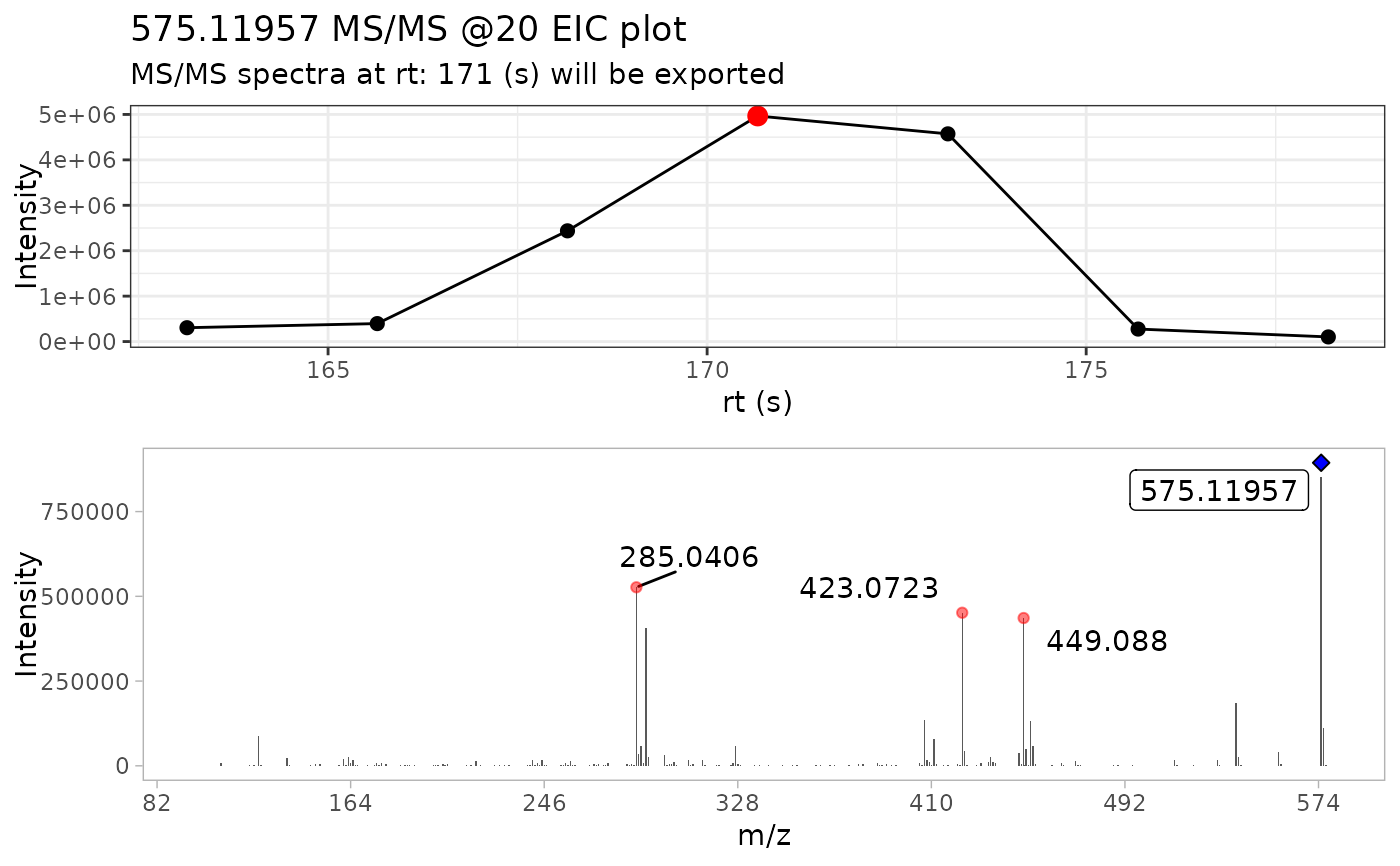 # Detecting masses with the normalized spectra and ions with
# intensities greater than 1%
ProcA2_detected <- detect_mass(ProcA2_ext,
normalize = TRUE, # Allow normalization
min_int = 1
) # 1% as minimum intensity
# Reading the metadata
metadata_file <- system.file("extdata",
"msp_metadata.csv",
package = "MS2extract"
)
metadata <- read.csv(metadata_file)
#> Warning: incomplete final line found by readTableHeader on '/home/runner/work/_temp/Library/MS2extract/extdata/msp_metadata.csv'
# Exporting msp file
write_msp(
spec = ProcA2_detected,
spec_metadata = metadata,
msp_name = "Procyanidin_A2"
)
#> • Filtering MS/MS scans for 20 CE
# Example with batch spectra ----
# Select the csv file name and path
batch_file <- system.file("extdata", "batch_read.csv",
package = "MS2extract"
)
# Read the data frame
batch_data <- read.csv(batch_file)
# File paths for Procyanidin A2 and Rutin
ProcA2_file <- system.file("extdata",
"ProcyanidinA2_neg_20eV.mzXML",
package = "MS2extract"
)
Rutin_file <- system.file("extdata",
"Rutin_neg_20eV.mzXML",
package = "MS2extract"
)
# Add file path - User should specified the file path -
batch_data$File <- c(ProcA2_file, Rutin_file)
# Checking batch_data data frame
batch_data
#> Name Formula Ionization_mode min_rt max_rt COLLISIONENERGY
#> 1 Procyanidin A2 C30H24O12 Negative 163 180 20 eV
#> 2 Rutin C27H30O16 Negative 162 171 20 eV
#> File
#> 1 /home/runner/work/_temp/Library/MS2extract/extdata/ProcyanidinA2_neg_20eV.mzXML
#> 2 /home/runner/work/_temp/Library/MS2extract/extdata/Rutin_neg_20eV.mzXML
# Reading batch metadata
metadata_msp_file <- system.file("extdata",
"batch_msp_metadata.csv",
package = "MS2extract"
)
metadata_msp <- read.csv(metadata_msp_file)
#> Warning: incomplete final line found by readTableHeader on '/home/runner/work/_temp/Library/MS2extract/extdata/batch_msp_metadata.csv'
# Using batch import to import multiple compounds
batch_compounds <- batch_import_mzxml(batch_data)
#>
#> ── Begining batch import ───────────────────────────────────────────────────────
#>
#> ── -- ──
#>
#> • Processing: ProcyanidinA2_neg_20eV.mzXML
#> • Found 1 CE value: 20
#> • Remember to match CE velues in spec_metadata when exporting your library
#> • m/z range given 10 ppm: 575.11376 and 575.12526
#> • Compound name: Procyanidin A2_Negative_20
#>
#> ── -- ──
#>
#> • Processing: Rutin_neg_20eV.mzXML
#> • Found 1 CE value: 20
#> • Remember to match CE velues in spec_metadata when exporting your library
#> • m/z range given 10 ppm: 609.14002 and 609.15221
#> • Compound name: Rutin_Negative_20
#>
#> ── End batch import ────────────────────────────────────────────────────────────
# Checking dimension by compound
# Procyanidin A2: 24249 ions
# Rutin: 22096 ions
purrr::map(batch_compounds, dim)
#> $`Procyanidin A2_Negative_20`
#> [1] 17829 6
#>
#> $Rutin_Negative_20
#> [1] 11475 6
#>
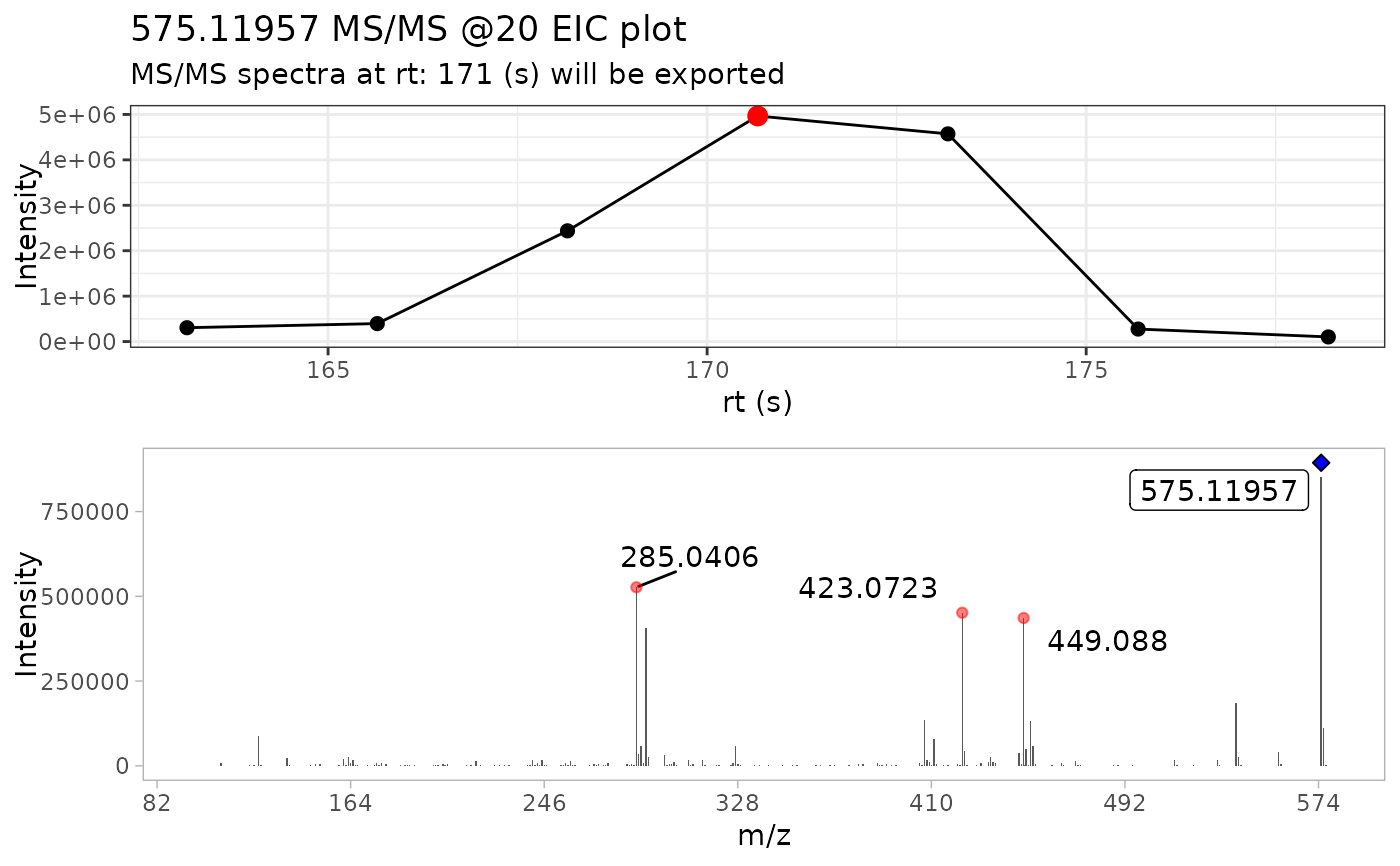
batch_extracted_compounds <- batch_extract_MS2(batch_compounds)
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Warning: `position_stack()` requires non-overlapping x intervals.
# Detecting masses with the normalized spectra and ions with
# intensities greater than 1%
ProcA2_detected <- detect_mass(ProcA2_ext,
normalize = TRUE, # Allow normalization
min_int = 1
) # 1% as minimum intensity
# Reading the metadata
metadata_file <- system.file("extdata",
"msp_metadata.csv",
package = "MS2extract"
)
metadata <- read.csv(metadata_file)
#> Warning: incomplete final line found by readTableHeader on '/home/runner/work/_temp/Library/MS2extract/extdata/msp_metadata.csv'
# Exporting msp file
write_msp(
spec = ProcA2_detected,
spec_metadata = metadata,
msp_name = "Procyanidin_A2"
)
#> • Filtering MS/MS scans for 20 CE
# Example with batch spectra ----
# Select the csv file name and path
batch_file <- system.file("extdata", "batch_read.csv",
package = "MS2extract"
)
# Read the data frame
batch_data <- read.csv(batch_file)
# File paths for Procyanidin A2 and Rutin
ProcA2_file <- system.file("extdata",
"ProcyanidinA2_neg_20eV.mzXML",
package = "MS2extract"
)
Rutin_file <- system.file("extdata",
"Rutin_neg_20eV.mzXML",
package = "MS2extract"
)
# Add file path - User should specified the file path -
batch_data$File <- c(ProcA2_file, Rutin_file)
# Checking batch_data data frame
batch_data
#> Name Formula Ionization_mode min_rt max_rt COLLISIONENERGY
#> 1 Procyanidin A2 C30H24O12 Negative 163 180 20 eV
#> 2 Rutin C27H30O16 Negative 162 171 20 eV
#> File
#> 1 /home/runner/work/_temp/Library/MS2extract/extdata/ProcyanidinA2_neg_20eV.mzXML
#> 2 /home/runner/work/_temp/Library/MS2extract/extdata/Rutin_neg_20eV.mzXML
# Reading batch metadata
metadata_msp_file <- system.file("extdata",
"batch_msp_metadata.csv",
package = "MS2extract"
)
metadata_msp <- read.csv(metadata_msp_file)
#> Warning: incomplete final line found by readTableHeader on '/home/runner/work/_temp/Library/MS2extract/extdata/batch_msp_metadata.csv'
# Using batch import to import multiple compounds
batch_compounds <- batch_import_mzxml(batch_data)
#>
#> ── Begining batch import ───────────────────────────────────────────────────────
#>
#> ── -- ──
#>
#> • Processing: ProcyanidinA2_neg_20eV.mzXML
#> • Found 1 CE value: 20
#> • Remember to match CE velues in spec_metadata when exporting your library
#> • m/z range given 10 ppm: 575.11376 and 575.12526
#> • Compound name: Procyanidin A2_Negative_20
#>
#> ── -- ──
#>
#> • Processing: Rutin_neg_20eV.mzXML
#> • Found 1 CE value: 20
#> • Remember to match CE velues in spec_metadata when exporting your library
#> • m/z range given 10 ppm: 609.14002 and 609.15221
#> • Compound name: Rutin_Negative_20
#>
#> ── End batch import ────────────────────────────────────────────────────────────
# Checking dimension by compound
# Procyanidin A2: 24249 ions
# Rutin: 22096 ions
purrr::map(batch_compounds, dim)
#> $`Procyanidin A2_Negative_20`
#> [1] 17829 6
#>
#> $Rutin_Negative_20
#> [1] 11475 6
#>
batch_extracted_compounds <- batch_extract_MS2(batch_compounds)
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Warning: `position_stack()` requires non-overlapping x intervals.
 #> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Warning: `position_stack()` requires non-overlapping x intervals.
#> Scale for x is already present.
#> Adding another scale for x, which will replace the existing scale.
#> Warning: `position_stack()` requires non-overlapping x intervals.
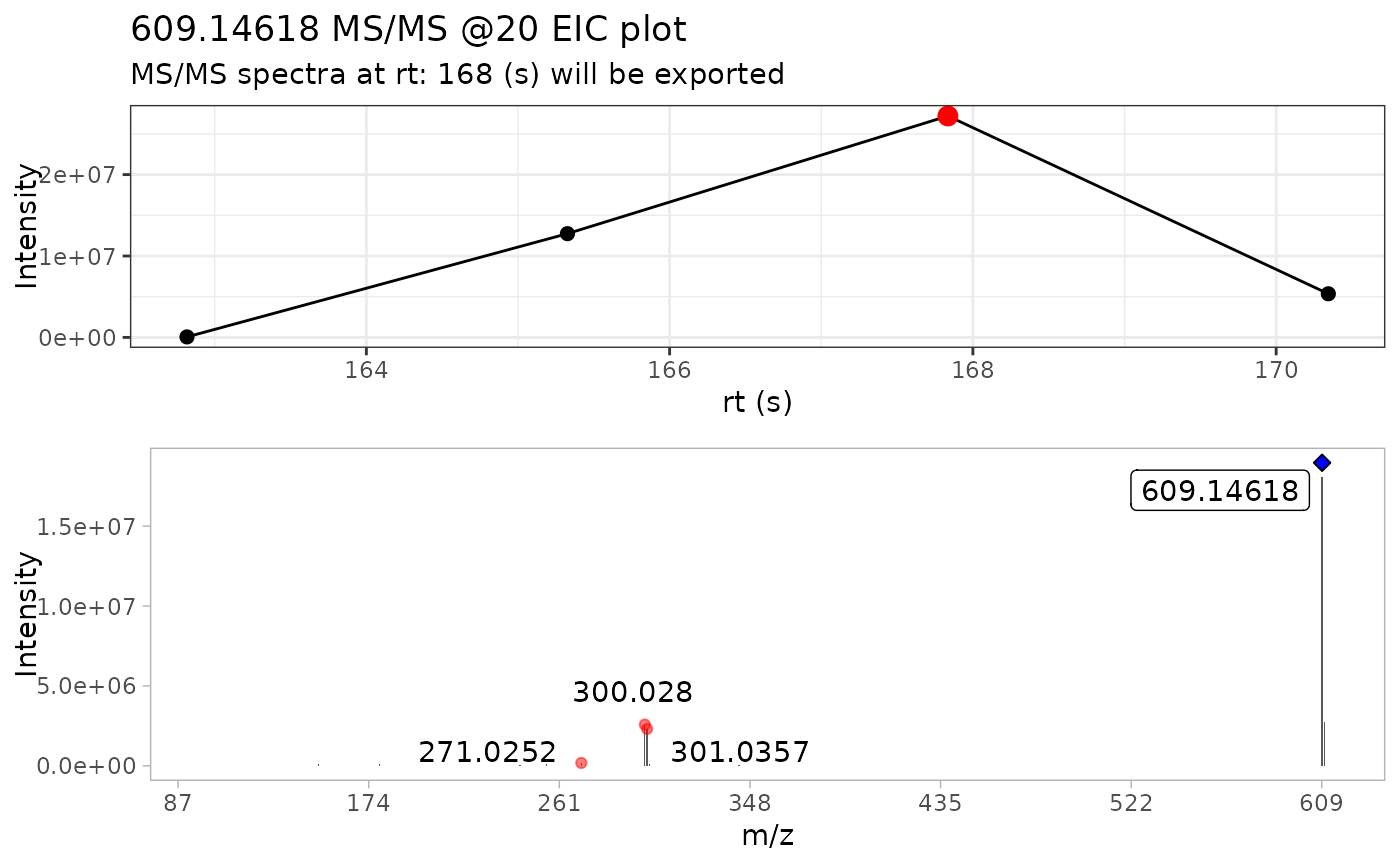 # Batch detect mass
batch_mass_detected <- batch_detect_mass(batch_extracted_compounds, # Compound list
normalize = TRUE, # Normalize
min_int = 1
) # Minimum intensity
# Checking dimension by compound
# Procyanidin A2: 107 ions
# Rutin: 12 ions
purrr::map(batch_mass_detected, dim)
#> $`Procyanidin A2_Negative_20`
#> [1] 38 6
#>
#> $Rutin_Negative_20
#> [1] 4 6
#>
# Writing msp file
write_msp(
spec = batch_mass_detected,
spec_metadata = metadata_msp,
msp_name = "ProcA2_Rutin_batch"
)
#> • Filtering MS/MS scans for 20 CE
#> • Filtering MS/MS scans for 20 CE
# Batch detect mass
batch_mass_detected <- batch_detect_mass(batch_extracted_compounds, # Compound list
normalize = TRUE, # Normalize
min_int = 1
) # Minimum intensity
# Checking dimension by compound
# Procyanidin A2: 107 ions
# Rutin: 12 ions
purrr::map(batch_mass_detected, dim)
#> $`Procyanidin A2_Negative_20`
#> [1] 38 6
#>
#> $Rutin_Negative_20
#> [1] 4 6
#>
# Writing msp file
write_msp(
spec = batch_mass_detected,
spec_metadata = metadata_msp,
msp_name = "ProcA2_Rutin_batch"
)
#> • Filtering MS/MS scans for 20 CE
#> • Filtering MS/MS scans for 20 CE